Input files
Suports calculations for a binary system \(A_{x}B_{1-x}\) or a pseudobinary system \(A_{x}B_{1-x}C\) , x=0~1.
input file I
The grand canonical takes into account the local compositional fluctuation and enables to predict the average value of the “property of interest” (PoI) for any composition and temperature. However, it requires the energy and PoI for all possible compositions and configurations, which is not practically feasible.
Therefore, P5Grand requires the PoI of feasibly large number of (not all) configurations, which are randomly generated at each possible composition.
The energy and property of each configuration can be obtained by diverse ways such as:
all by DFT calculations
by machine learning based on some DFT data
by empirical-based data
For the free energy mode and the phase diagram mode, it consists of two columns and many rows. Each column presents the composition (x) and the energy (Eσ) of a certain configuration (σ) while each row corresponds to each configuration.
0 -8.675196
0.03125 -8.700504
0.0625 -8.725803
0.0625 -8.726524
0.0625 -8.726524
...
For the PoI mode, it consists of three columns and many rows. Each column presents x, Eσ and PoI of a certain configuration.
0.0 -9.189511 0.713310
0.03125 -9.211485 0.676563
0.0625 -9.234508 0.691304
0.0625 -9.233217 0.660815
0.0625 -9.234138 0.672744
...
Each row indicates a configuration. The first and the second column is the composition and energy of a configuration. The third column is PoI like bandgap of a configuration.
The composition of a configuration (the first column) should be within 0~1.
The unit of the energy of a configuration (the second column) can be either eV/[mixing atoms] or J/[mixing atoms].
In this document, the name of input file is referred to as CEL.log, which is the default name of input file to be read in P5Grand.
input file II
The grand canonical requires the strain energy induced by local compositional fluctuation. In this document, the strain induced by local compositional fluctuation is referred to as local strain.
P5Grand approximates the local strain energy to Birch-Murnaghan equation of state using several parameters. As a default, P5Grand requires a file which contains four parameters.
\(B_0V_0 \ \ \ B'_0 \ \ \ 1 \ \ \ \frac{V_0(x=1)}{V_0(x=0)}\)
, where V0 and V are free relaxed volume and strained volume, B0 and B’0 are bulk modulus and its derivative.
The first and second values are V0 B0 and B’0, respectively. Fourth value over third value should be the V0(x=1) divided by V0(x=0), which limits V/V0 to be calculated.
The fitting equations can be manually modified by editing the local_strain.py module.
Note
The unit of strain energy should be same to the unit of energy in CEL.log, either eV/[mixing atoms] or J/[mixing atoms].
[mixing atoms] indicates the number of atoms in microstates. For a pseudobinary system of GaAsxSb1-xas an example, a 2x2x2 zincblende supercell is composed of 32 cations and 32 anions. In this case, the number of mixing atoms is 32.
tool/extract_strain
P5Grand provides tool/extract_strain.py script to generate a file containing fitting parameters.
tool/extract_strain.py script extracts parameters for local strain from the OUTCAR of VASP, and automatically write them in a file named BM_constant.dat.
For each configuration, directories should be treed as below.
configuration
├── unstrained
│ └── OUTCAR
├── 0_strained
│ └── OUTCAR
├── .
│ .
├── .
│ .
└── n_strained
└── OUTCAR
Note
Directory named unstrained which calculates freely relaxed state is essential.
Other directories involves the calculation results for strained state.
Every lowest subdirectory should involve OUTCAR.
Overall directory tree is follows.
strain
├── x_equal_0_and_1_must_be_included
│ ├── 0
│ │ ├── unstrained
│ │ │ └── OUTCAR
│ │ ├── 0_strained
│ │ │ └── OUTCAR
│ │ ├── .
│ │ │ .
│ │ ├── .
│ │ │ .
│ │ └── n_strained
│ │ └── OUTCAR
│ └── 1
│ ├── unstrained
│ │ └── OUTCAR
│ ├── 0_strained
│ │ └── OUTCAR
│ ├── .
│ │ .
│ ├── .
│ │ .
│ └── n_strained
│ └── OUTCAR
├── configuration_1
│ ├── unstrained
│ │ └── OUTCAR
│ ├── 0_strained
│ │ └── OUTCAR
│ ├── .
│ │ .
│ ├── .
│ │ .
│ └── n_strained
│ └── OUTCAR
├── configuration_2
│ ├── unstrained
│ │ └── OUTCAR
│ ├── 0_strained
│ │ └── OUTCAR
│ ├── .
│ │ .
│ ├── .
│ │ .
│ └── n_strained
│ └── OUTCAR
├── .
│ .
├── .
│ .
└── configuration_n
├── unstrained
│ └── OUTCAR
├── 0_strained
│ └── OUTCAR
├── .
│ .
├── .
│ .
└── n_strained
└── OUTCAR
Note
Directories for x=0 and x=1 should be included.
Enter the below command in strain directory to extract the parameters for local strain from the OUTCAR of VASP.
$ python /dir/to/P5Grand/tool/extract_strain.py atomA atomB
An example of this command is provided at example/strain for the case of GaAsxSb1-x. For this example, the command is:
$ python /dir/to/P5Grand/tool/extract_strain.py As Sb
Then, you can get BM_constant.dat file and fitting graph as follows.
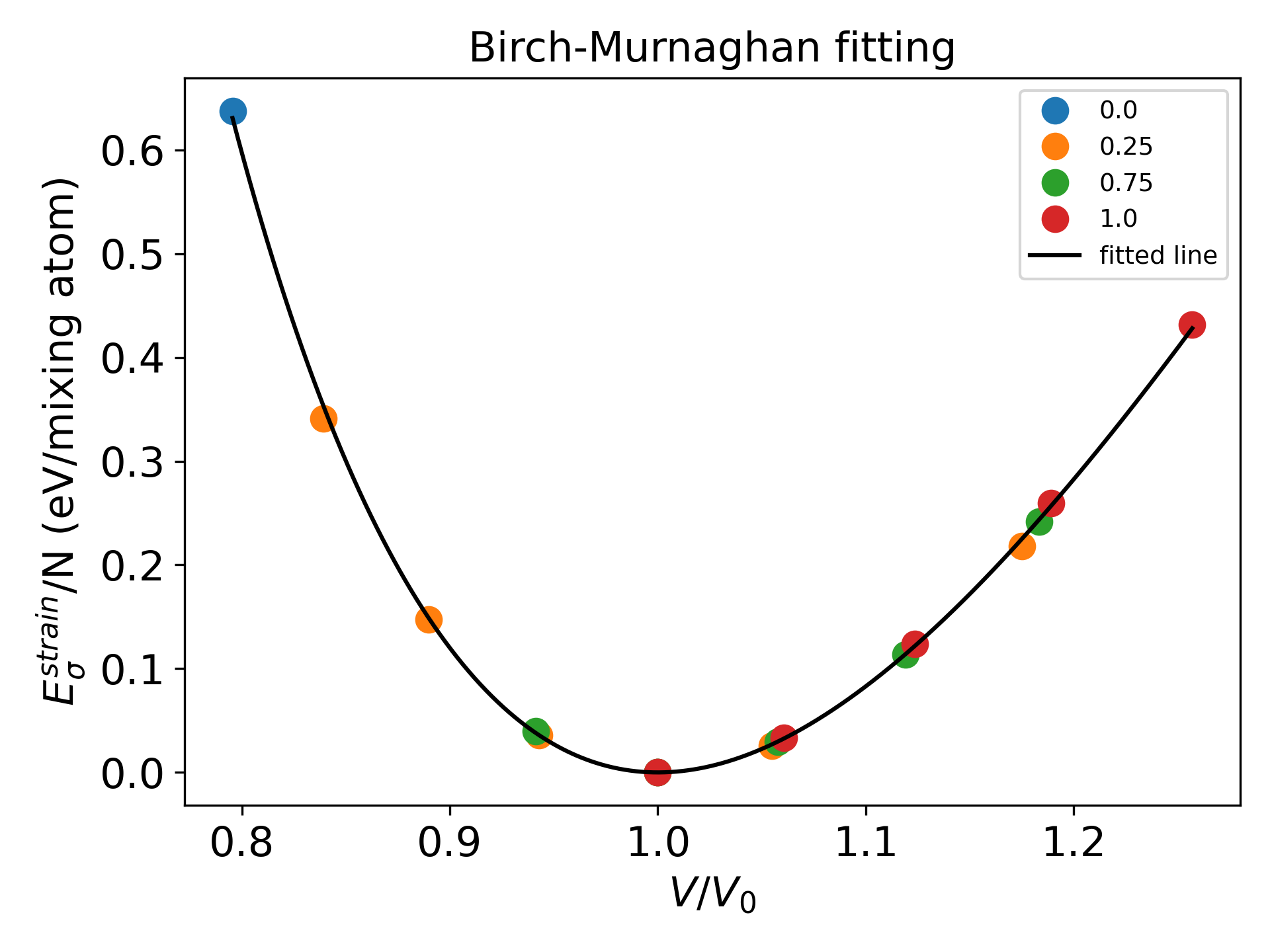
The fitting equation can be manually modified by editing the tool/extract_strain.py script, and will be updated to account for the composition-dependent cases.